Are we closer to the truth? Parkinson’s disease variability across populations tells us something about its genetic contributors
Disclaimer: This web page was produced as an assignment for an undergraduate course at Davidson College.
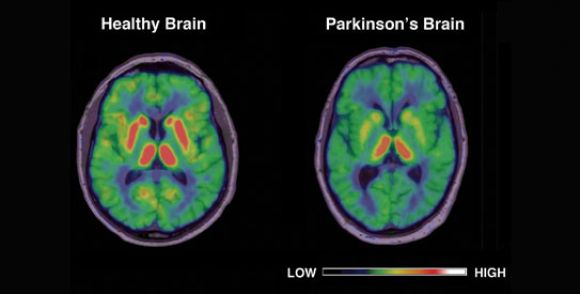
Image courtesy of Marios Politis / AAAS
Parkinson’s Disease is the fastest moving neurodegenerative disease that affects 8.5 million people in the world (WHO 2022). Researches have been keen to find the underlying mechanisms for this devastating disease. In a study conducted by (Kim, J.J et al. 2023), they aimed to identify the genes involved in Parkinson’s Disease, and create the largest and most diverse set of participants of Parkinson’s Disease genomics.
Parkinson’s Disease is caused by neural deterioration in a brain region called Substantia Nigra, which produces dopamine, a neurotransmitter that is responsible for movement and reward. As well as the presence of Lewy Bodies. As a result of the neuronal deterioration of Subantia Nigra, people with Parkinson’s Disease exhibit symptoms of loss of movement coordination, balance, and other psychological issues such as depression (Zafar S et al. 2023). Due to Parkinson’s Disease inheritance patterns, researchers aimed to look at PD-specific genes to try to find a possible Single Nucleotide Polymorphism (SNP) related to the disease.
An SNP is a type of mutation whereby only one of the coding letters (A, T, C, G) change into another, causing a divergence from the typical protein that is encoded by a 3-letter combination (called the codon). When the protein that is encoded does not match the protein that was supposed to be encoded, it leads to a change in a phenotypic – or apparent- trait. Identifying SNPs and associating them to a certain trait is crucial to identifying genes that are contributing to the disease. Because of the variation between individuals from different ancestries in their SNPs and how these affect phenotypes, it is important for any genomic research to include large sets of data to represent all populations possible, and be able to treat and/or manage the disease accurately.
Kim J.J et al. conducted a Genome-Wide Association Study (GWAS) which studies specific genetic variations and its relation to different traits by comparing genomes from different individuals (Uffelmann et al. 2021). In their 2023 study, they compiled genomic data from almost 50,000 individuals with PD, more than 18,000 proxy cases (caregivers reporting on behalf of individuals with PD), and almost 2,500,000 control individuals. The study gathered this data from individuals from four different ancestry groups; European, East Asian, Latin American, and African ancestries.
The researchers focused on finding the SNPs that are shared between individuals from different ancestries, using multiple levels of analysis. First, they conducted a linkage disequilibrium (LD) score regression, that looks at the non-random association between genes. When LD score is low, it means there is little association between the genes (Teo Yik Y et al. 2010). The study found there was a low LD within ancestries, which means there is no overlapping samples between the different populations and a high level of confidence interval. This step was an important one to ensure that the data was ready to be investigated.
Second, they performed a random-effects model to detect for homogeneous allelic effects, and a meta-regression multi-ethnic genetic association (MR-MEGA) to detect heterogenous effects across different populations. By combining the results from both models, the researchers found 12 novel PD risk loci, and 66 independent risk loci that were previously known. Of these novel 12 loci, 9 were found to have homogenous effects, while 3 had heterogenous effects across different populations. The study further confirmed that these genes were located in brain regions that are involved in elevated PD risk. MTF2 and PPP6R2 loci contain the genes TMED5 and PPP6R2 both which code for protein that are involved in the localization of Golgi body and form part of the vesicular transport pathway respectively, implicating their PD pathogenesis.
On another hand, some genes that were found in suggestive loci were only found in Latin American and African populations. JAK1 is an important protein implicated in cytokine and inflammatory signaling, the variants of this protein contribute to autoimmune diseases, such as multiple sclerosis. HS1BP3 has been implicated in essential tremor. The researches suggest that if these two genes are confirmed, they would support the role of inflammation in PD. Further, the gene ontology conducted found new pathways that might be relevant to PD. Some of these pathways include related to mitochondria vesicles, tau protein and immune cells. Some of these pathways had not been detected before in the all-European study.
The researchers acknowledge the lack of true representation in their data despite their efforts, as African population representation is only 0.5% of the PD cases, while the European cases remain the majority of all cases studied taking 80% of the GWAS. The research might have oversold the idea of equitable representation, and in a study implicating a fatal disease it is crucial for researchers to involve as many individuals as possible from different backgrounds; this becomes even more important after finding two genes, JAK1 and HS1BP3 that are unique to certain populations. The study does a good job in going from a populations approach to a molecular approach, identifying and targeting specific brain tissues related to the appearance of the pathogen, as well as identifying the new putative loci. This paper opens up new venues for research in rat models by creating knock-out genes to test for Loss-of-Function and determine whether the genes are involved causally.
References
- Kim, J.J., Vitale, D., Otani, D.V. et al. Multi-ancestry genome-wide association meta-analysis of Parkinson’s disease. Nat Genet 56, 27–36 (2024). https://doi.org/10.1038/s41588-023-01584-8
- Teo, Yik Y et al. “Power consequences of linkage disequilibrium variation between populations.” Genetic epidemiology vol. 33,2 (2009): 128-35. doi:10.1002/gepi.20366
- Uffelmann, E., Huang, Q.Q., Munung, N.S. et al. Genome-wide association studies. Nat Rev Methods Primers 1, 59 (2021). https://doi.org/10.1038/s43586-021-00056-9
- Zafar S, Yaddanapudi SS. Parkinson Disease. [Updated 2023 Aug 7].
- World Health Organization Launch of WHO’s Parkinson disease technical brief (2022)
Iasa Hatem
iahatem@davidson.edu
© Copyright 2022 Department of Biology, Davidson College, Davidson, NC 28036
One of the major philanthropic events that my high school organized was an ALS run/walk fundraiser. This event taught me a lot, and while I know that Parkinson’s is its own distinct disease, their similarities drew me to read your article. I liked the way that you detailed the overall methodology of your study, and found various parallels to my own. You did a great job in detailing what we stand to gain from larger population representation in genomics studies, and you were not afraid to call out your own article for failing to admit their shortcomings.
My grandmother has PD which is what led me to read this post. I have grown up hearing about the debate surrounding the cause of Parkinson’s, and whether it is genetic or environmental. Personally, I believe environment plays a larger role but I also believe there could be genetic factors that make someone more vulnerable. I think this work to identify relevant genes is very important, especially as someone who has a family history of PD. I would love for there to be a form of genetic testing that could tell family members if they have inherited an at risk gene, as there is for Alzheimers and breast cancer.