Written By: Porter Alston | poalston@davidson.edu
This web page was produced as an assignment for an undergraduate course at Davidson College.
Comparing the performance of traditional short-read DNA sequencing with long-read sequencing reveals significant differences in the abilities of these technologies to accurately sequence complex, medically relevant gene variants. The results show that long-reads produce the most accurate results for both small and large variants, improving as software becomes optimized for variant detection during sequence alignment.
The field of genomics is rapidly developing, with innovations to procedure, technology, and access to resources increasing exponentially each year. The backbone of genomics is the ability to interact with DNA and read the base pairs which make up all of life’s chromosomes, also known as sequencing. The technology of sequencing requires the amplification of genetic material in a chain reaction which generates a sample of variable-length DNA strands, or reads, which are big enough to be sorted and ordered with relative accuracy. Currently, there are two types of sequencing methodologies: short-read sequencing and long-read sequencing. Illumina sequencing is a leading technology for short-read sequencing, providing a relatively cheap alternative to the long-read sequencing technologies produced by Oxford Nanopore Technology and Pacific Biosciences. However, recent studies aim to compare short-read and long-read sequence alignment using newer genomic data and software, demonstrating the utility of long-read sequencing in genomic studies related to health.
One of the earliest forms of sequencing was created and optimized by Illumina in 2008.1 Illumina uses sequencing by synthesis, a process during which fluorescent markers help read a single-chain deoxyribonucleic acid (DNA) strand. The fluorescent group indicates which nucleotides are present at each position, allowing for sequential analysis of each position. Short-read sequencing using Illumina technology typically generates a read length of 50-300 base pairs.1
Next-generation sequencing, or long-read sequencing, gained popularity with its introduction to the field of genomics in 2014.1 The first commercially available long-read technology was from Pacific Biosciences. The method started with continuous long-reads which produced a high error rate. However, developments in the procedure have led to high-fidelity reads with an error rate lower than 1%.1 Long-read technology from Pacific Biosciences produces a typical read of 15 to 20 kilobase pairs. Shortly after Pacific Biosciences, Oxford Nanopore Technologies began nanopore sequencing, a long-read sequencing technology which provides reads up to 4 megabase pairs. While these reads used to suffer from a higher sequencing error, innovations in double-strand sequencing are reducing error rates to levels seen in high-fidelity reads.1
The accuracy of sequence reads are imperative to uncovering the correlation between genes and phenotypic outcomes. For this reason, Mahmoud et al. conducted an experiment to compare the accuracy in detection of genetic mutations in medically correlated genes between each of the sequencing methods.2 In order to accurately detect a gene variant, or “recall” a mutation, each part of the genetic sequence must be read multiple times, a tool known as coverage. They determined a short-read Illumina coverage between 29.76× and 32.50× provides sufficient accuracy while they obtained an average coverage of 45.29× for long-reads using Oxford Nanopore Technologies and 35.70× for Pacific BioSciences (Figure 1).
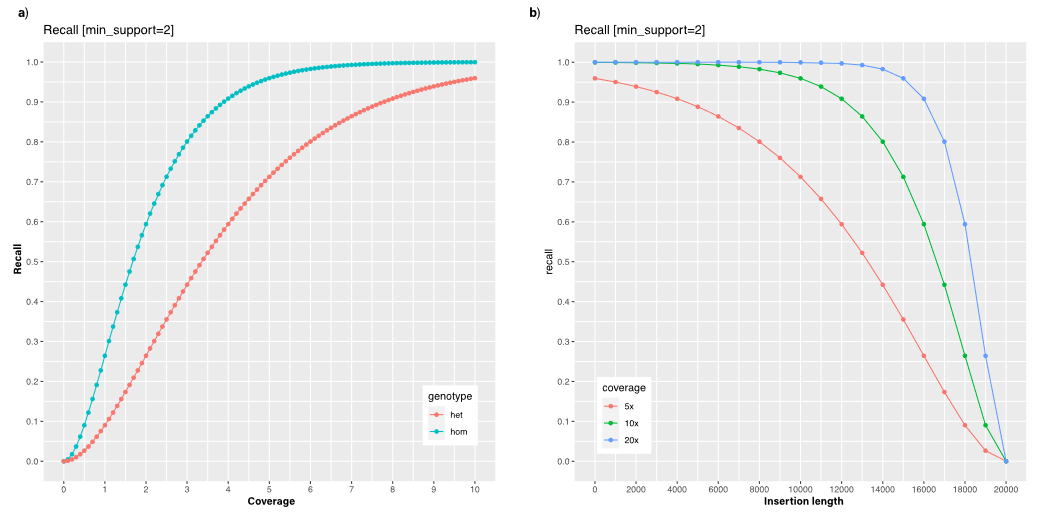
Figure 1: Assessment of variant calling as a function of coverage and insert size. These graphs depict general trends for sequencing technology with regards to coverage’s effect on mutational variant recall. Regardless of the sequencing technology, coverage correlates with recall using the same function. a) A graph depicting the effects of optimal coverage on recall of mutational variants based on the genotype of alleles. Both heterozygous and homozygous genes follow the same trend in recall, with increased coverage correlated with increased recall of mutational variants. b) A graph depicting the effects of mutational insertion length on recall of structural mutations. As the average size of a mutation increases, the recall of mutational variants tends to decrease.
Once the coverage needed to optimize variant detection was determined, researchers began using publicly available genomic data to compare the accuracy of sequence reads between each technology. As a control, researchers used data from the National Institute of Health’s All of Us program, in which multiple cell lines for each sample have already been sequenced and cataloged using only short-read sequencing. Researchers then resequenced samples from the All of Us program using short and long-read sequencing technology as well as using different software programs for sequence alignment. This resulted in new entire genome sequences for multiple samples in the All of Us genome, each done with both methods of sequencing and each with a control comparison from the original All of Us sample set. From here, researchers narrowed their window of interest in the genome to areas correlated to medical disease and poor health outcomes in order to see which method better detected correlated mutations.
For mutations of size less than 50 base pairs, long and short-read technologies agreed on approximately 67.55% of mutations with the majority of unique mutations found in short-read sequences to be errors. For mutation variants greater than 50 base pairs, concordance between the three technologies is approximately 22.00%. However, long-read technologies agreed on 53.86% of all longer mutations with 31.90% of longer mutations remaining undetected in short-reads. Interestingly, 15% of all longer mutations were found exclusively by short-read technology.
For further analysis, 4,641 medically relevant genes mapped in the All of Us dataset were analyzed and compared between sequencing technologies. Comparison shows the greatest lack in coverage of these genes by short-read sequencing, while Oxford Nanopore Technologies covers all genes followed closely in coverage by Pacific Biosciences. For identification of all medically relevant mutations of interest, researchers found a consistently higher accuracy in both long-read technologies when compared to short-reads. The same was true for coverage, demonstrating a direct correlation between the ability of long-sequence reads to detect mutations as well as identify medically relevant genes.
Pacific Biosciences outperformed all other technologies in both precision and recall. Technology comparison demonstrated that there are only a few challenging genes for which implementation of all three technologies still results in lack of coverage. Comparison of short and long-read sequencing gives reason for continued development of genomic population data sequenced with long-reads. Currently, the primary downside to long-reads is a slight reduction in accuracy across small mutations. Accuracy can be improved through continued use of short-read sequencing in combination with long-read technologies as well as through development of computational software for recall of mutations. Ultimately, as the field of genomics expands, long-read sequencing is growing our understanding of genetic mutations and their effects on health. With more research and sequencing using long-reads, the true variation within the genome can be better described and cataloged, revealing genetic correlations to disease which would be undetectable by short-read technology.
References:
(1)
Heather, J. M.; Chain, B. The Sequence of Sequencers: The History of Sequencing DNA. Genomics 2016, 107 (1), 1. https://doi.org/10.1016/j.ygeno.2015.11.003.
(2)
Mahmoud, M.; Huang, Y.; Garimella, K.; Audano, P. A.; Wan, W.; Prasad, N.; Handsaker, R. E.; Hall, S.; Pionzio, A.; Schatz, M. C.; Talkowski, M. E.; Eichler, E. E.; Levy, S. E.; Sedlazeck, F. J. Utility of Long-Read Sequencing for All of Us. Nat Commun 2024, 15 (2), 837. https://doi.org/10.1038/s41467-024-44804-3.
© Copyright 2024 Department of Biology, Davidson College, Davidson, NC 28036.
I think you did a great job describing the differences, pros and cons of both types of sequencing. Moreover, the experimental design was well summarized and described. It was an interesting approach that the researchers took to re sequence genomes with both technologies and compare their efficacy. I think It is very interesting to see how long reads outperformed short reads, especially thinking of how they are known for being less precise than short read sequencing.